Machine-Learned Collective Variables and Their Associated Free Energy Surfaces
The rapid convergence of machine learning with enhanced sampling and free-energy methodologies marks a genuine paradigm shift: starting with the automated design of collective variables, machine learning is now reshaping how we represent, explore, and interpret molecular free-energy landscapes-blurring the boundaries between sampling, bias construction, and thermodynamic inference, and signalling a revolution in the way atomistic simulations generate physical insight. Building on the theoretical and computational foundations discussed in the previous sections, this new generation of approaches leverages data-driven representations to couple learning and sampling in closed loops, enabling adaptive exploration of high-dimensional configuration spaces. Here, we focus on some key aspects relevant to the computation and interpretation of free-energy surfaces. For a broader, more comprehensive perspective on the ongoing integration of machine learning, enhanced sampling, and free energy methods, we refer the reader to the extensive overviews by Noé et al. Noé et al., 2020, Mehdi et al. Mehdi et al., 2024, and Zhu et al. Zhu et al., 2025.
Machine-Learning CVs¶
Traditionally, collective variables (CVs) have been designed from physical intuition, for example, distances in ion-pair association, torsional angles in peptide isomerization, or bond-order parameters in crystallization. Such handcrafted descriptors have been instrumental in molecular simulations for decades, enabling the projection of complex dynamics onto interpretable coordinates (see Section sec:CVs). Yet, they are rarely optimal. In high-dimensional systems, where the relevant slow modes arise from nonlinear couplings of many atomic degrees of freedom, physically inspired CVs may fail to distinguish metastable states or to capture the true kinetic bottlenecks of phase-space exploration. This realization has prompted the development of machine-learned collective variables (MLCVs), which employ data-driven algorithms to infer optimal low-dimensional representations directly from simulation trajectories Gökdemir & Rydzewski, 2025Desgranges & Delhommelle, 2025Neha et al., 2022. The field of MLCVs encompasses a spectrum of methodologies that can be broadly grouped by the learning principle they adopt. Unsupervised approaches - including principal component analysis Garcı́a, 1992Amadei et al., 1993Hegger et al., 2007Wetzel, 2017, diffusion maps Ferguson et al., 2011Preto & Clementi, 2014, sketch-map Ceriotti et al., 2011Tribello et al., 2012, and autoencoders Chen & Ferguson, 2018Wehmeyer & Noé, 2018Lemke & Peter, 2019-learn low-dimensional manifolds that preserve the variance or geometric structure of the high-dimensional trajectory data. These methods are effective for capturing dominant structural fluctuations, such as protein conformational changes or order--disorder transitions in solids. Still, they do not guarantee the recovery of dynamical slow modes. In contrast, variational and supervised methods explicitly target dynamical relevance. The variational approach to conformational dynamics (VAC) Noé & Nuske, 2013Nuske et al., 2014, defines an optimal reaction coordinate as the mapping that maximizes the time-lagged autocorrelation of the projected dynamics---equivalently, the leading eigenfunction of the transfer operator. Implementations such as time-lagged independent component analysis (TICA) Pérez-Hernández et al., 2013, VAMPnets Mardt et al., 2018, and state-free reversible VAMPnets (SRV) Chen et al., 2019 learn nonlinear transformations that approximate these slow eigenmodes, often in the form of neural-network embeddings. Closely related are information-bottleneck formulations, such as RAVE (reweighted autoencoded variational Bayes for enhanced sampling) Ribeiro et al., 2018, SPIB Wang et al., 2019 and variational dynamic encoders (VDE) Hernández et al., 2018, which train neural networks to identify minimally complex yet maximally predictive latent variables, thus approximating the committor function (see the dedicated box above). Complementary to these are discriminant-based approaches that use labeled metastable states: linear discriminant analysis (LDA) and its harmonic variant (HLDA) construct transparent, differentiable linear CVs that maximize between-state separation Mendels et al., 2018Piccini et al., 2018. Their nonlinear extensions, Deep-LDA and Deep-TDA, replace the linear map with a neural network, yielding smooth, expressive CVs that integrate seamlessly with biased enhanced sampling methods such as US, MetaD, VES, and OPES for free-energy reconstruction Bonati et al., 2020Trizio & Parrinello, 2021. These algorithms have found widespread applications across various fields, including ligand binding Wang et al., 2019, conformational transitions Hernández et al., 2018Mardt et al., 2018Chen et al., 2019, self-assembly Jung et al., 2023Boninsegna et al., 2018, and phase transformations Finney & Salvalaglio, 2023.
A recent frontier in the construction of MLCVs is the use of graph neural networks (GNNs) and geometric deep learning architectures, which natively encode the fundamental symmetries of molecular systems - translation, rotation, and permutation invariance of identical atoms. By representing atomic environments as graphs with nodes (atoms) and edges (interactions), these architectures eliminate the need for explicit handcrafted descriptors such as symmetry functions or Steinhardt order parameters Dietrich et al., 2023Zhang et al., 2024. Two recent examples demonstrate the potential of this approach. In Dietrich et al. Dietrich et al., 2023, graph-based models were trained to approximate nucleation order parameters in colloidal and metallic systems. The learned variables reproduced the behaviour of conventional -based crystallinity measures, but with an order-of-magnitude computational speedup, enabling on-the-fly biasing in umbrella sampling and metadynamics. Similarly, Zhang et al. Zhang et al., 2024 introduced descriptor-free collective variables from geometric graph neural networks, extending the concept to molecular systems and demonstrating how equivariant layers can learn rotationally consistent embeddings of atomic environments without predefined order parameters.
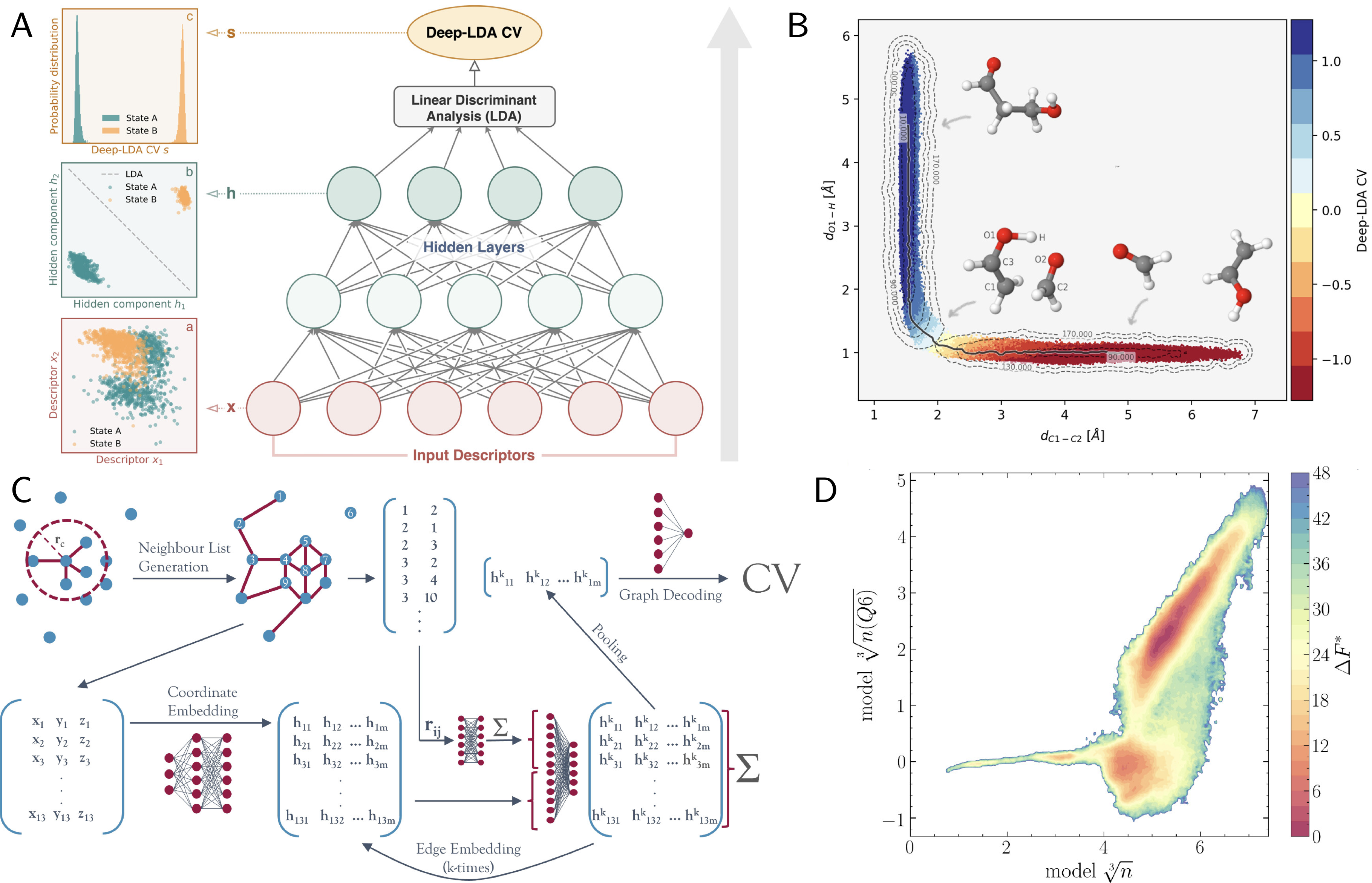
Figure 1:Data-driven (CVs) derived from deep learning. A,B. Deep-LDA Bonati et al., 2020 combines nonlinear neural-network transformations of physical descriptors with linear discriminant analysis to extract a one-dimensional CV that best separates metastable basins, yielding committor-like behavior (panel B) and interpretable feature rankings. C,D. Graph neural-network (GNN) CVs Dietrich et al., 2023 operate directly on molecular graphs built from atomic coordinates, aggregating local environments through message-passing layers and pooling to produce permutation-invariant, differentiable CVs transferable across system sizes and materials. These CVs can be deployed to carry out biased sampling with adaptive methods and thus compute FES, as seen in panel C, associated with the two-step nucleation of a colloidal system.
The natural complement to MLCVs is their integration with enhanced sampling methods (see section sec:Computing), where CVs serve as biasing coordinates to accelerate the simulation of rare events. Machine learning can be used both to discover effective CVs and to mitigate bias potential in real time. Iterative schemes such as active enhanced sampling alternate between sampling and learning, progressively refining the CV toward a dynamically optimal coordinate Ribeiro et al., 2018Ray et al., 2023Trizio et al., 2025. Such approaches are often developed in three main steps: (i) learn a low-dimensional representation of slow dynamics, (ii) deploy a given learnt representation as a biasing coordinate in MetaD/VES/OPES/ABF (see Section sec:Computing), and (iii) periodically re-train or fine-tune the model on newly sampled configurations. This iterative workflow, exemplified by, i.e. RAVE Ribeiro et al., 2018, is highlighted and discussed in detail across recent reviews Noé et al., 2020Mehdi et al., 2024Zhu et al., 2025 and case studies, where the bias is either optimized variationally Valsson & Parrinello, 2014Bonati et al., 2019 or built adaptively from sampling statistics.
Machine-Learning the Committor¶
The accurate sampling and learning of the committor in high-dimensional molecular systems remains an area of active research, uniting developments in enhanced-sampling algorithms, transition-path theory and machine-learning representations to make the “ideal” reaction coordinate a practical and learnable object. In the variational approach presented by Kang et al. Kang et al., 2024 and Trizio et al. Trizio et al., 2025, the committor is represented as a differentiable model , where is a neural-network, function of physically motivated descriptors. The parameters of the neural network, , are optimized by maximizing the consistency between predicted and observed transition outcomes. This strategy generalizes the likelihood maximization of Peters and Trout Peters & Trout, 2006, and directly yields a smooth, differentiable approximation to the committor that can be analyzed, differentiated, and even symbolically regressed to human-interpretable forms. Applying a variational principle allows this problem to be reformulated in terms of the Kolmogorov functional:
whose minimization under boundary conditions and yields the committor function satisfying the Kolmogorov equation for overdamped dynamics Trizio et al., 2025.
This principle defines the Kolmogorov ensemble, in which configurations are sampled with probability , and where the committor-dependent bias stabilizes configurations belonging to the transition-state region Kang et al., 2024. Using this framework, Trizio et al. demonstrated that the learned approximation of the committor can be used not only to characterize the transition-state ensemble but also to drive enhanced sampling by coupling the Kolmogorov bias with on-the-fly probability enhanced sampling (OPES)Kang et al., 2024Trizio et al., 2025. In this extended formulation, the pre-activation of the neural network trained to yield the committor serves as a smooth CV that acts as a proxy for the committor itself and enables the exploration of metastable basins and transition states within a single self-consistent workflow. The resulting probability-based enhanced sampling approach, was used to model processes ranging from protein folding to ligand binding, accurately reproducing free-energy surfaces and reactive pathways while retaining interpretability and physical transparency @ Trizio et al., 2025.
FESs and sampling with MLCVs: some cautionary tales¶
As discussed above, MLCVs hold remarkable potential to complement the definition and calculation of useful FESs. Projecting onto MLCV spaces, however, raises subtle issues. As highlighted in Ref. Dietrich & Salvalaglio, 2025, the mapping , when obtained through an MLCV is not uniquely defined: different neural network trainings with identical architectures and hyperparameters can lead to different embeddings, altering the Jacobian matrix . Given that
The shape of can vary across training instances even when all models capture the same metastable states. This non-reproducibility problem is specific to machine-learned CVs and is absent in physically defined variables. An effective solution is the adoption of an alternative definition of the FES, common to applications in computational kinetics, i.e., the gauge-invariant or Geometric FES Hartmann & Schütte, 2007Hartmann et al., 2011:
where indicates the integral on the hypersurface defined by the level-set of all the configurations degenerate in , and the infinitesimal element of such hypersurface. This effectively removes the explicit Jacobian dependence of . Moreover, is invariant under any monotonic transformation of the CVs, ensuring that the levels of free energy minima, barriers, and saddle points are consistent across different training runs. This gauge invariance makes the natural framework for comparing FESs obtained from independently trained instances of MLCVs with the same architecture, and it favors comparisons across architectures parameterized with different hyperparameters. Another area where the application of MLCVs requires care is the reproducibility of sampling efficiency in biased simulations, since variability in the Jacobian arising from stochastic training and hyperparameter choices can lead to unreproducible biasing forces - including spikes or vanishing gradients - across different training instances (see Fig. fig:MLCVs_and_gradientsb) Dietrich & Salvalaglio, 2025. Building on the concept of Geometric FES discussed above, Ref. Dietrich & Salvalaglio, 2025 proposes gradient normalization as a simple and effective approach to mitigate this issue.
Machine-Learning-Enhanced Transition-Path Sampling¶
Machine learning is reshaping transition-path sampling (TPS) by enabling adaptive discovery of reactive trajectories in complex systems. Recent approaches merge ML inference with path-sampling algorithms, allowing the committor, transition paths, and even full path ensembles to be learned directly from data. Bolhuis and co-workers developed AIMMD Lazzeri et al., 2023, which trains a surrogate model for adaptive reweighting and importance sampling in trajectory space. Applied to chignolin folding, it reproduces free energies, rates, and mechanisms at a fraction of the cost of conventional TPS by learning the committor self-consistently. Jung et al. Jung et al., 2023 introduced an autonomous ML-TPS framework that iteratively trains a neural-network committor from shooting outcomes, refocusing sampling near transition states. Tested on ion association, hydrate nucleation, polymer folding, and membrane-protein assembly, it yields accurate rates and interpretable mechanisms without predefined reaction coordinates. Chipot and co-workers Chen et al., 2023 implemented the variational principle of transition-path theory using neural networks, inspiring later committor-consistent schemes such as PCCANN by Megías et al., which minimizes the finite-time-lag committor correlation function , to learn dominant transition tubes. Iteratively coupling biased MD sampling and committor retraining, PCCANN identifies committor-consistent pathways and multiple competing channels, demonstrated for NANMA isomerization and chignolin folding. Together, these studies mark a shift from fixed-bias sampling to data-driven path ensembles, in which machine learning reuses trajectory data, reduces costs, and generalizes across systems---laying the groundwork for future generative or diffusion-based transition-path theories.
- Noé, F., Tkatchenko, A., Müller, K.-R., & Clementi, C. (2020). Machine Learning for Molecular Simulation. Ann. Rev. Phys. Chem., 71(1), 361–390. 10.1146/annurev-physchem-042018-052331
- Mehdi, S., Smith, Z., Herron, L., Zou, Z., & Tiwary, P. (2024). Enhanced Sampling with Machine Learning. Ann. Rev. Phys. Chem., 75(2024), 347–370. 10.1146/annurev-physchem-083122-125941
- Zhu, K., Trizio, E., Zhang, J., Hu, R., Jiang, L., Hou, T., & Bonati, L. (2025). Enhanced Sampling in the Age of Machine Learning: Algorithms and Applications. ArXiv Prepr. ArXiv250904291. 10.1021/acs.chemrev.5c00700
- Gökdemir, T., & Rydzewski, J. (2025). Machine Learning of Slow Collective Variables and Enhanced Sampling via Spatial Techniques. Chem. Phys. Rev., 6(1). 10.32388/70q2ov
- Desgranges, C., & Delhommelle, J. (2025). Deciphering the Complexities of Crystalline State (s) with Molecular Simulations. Commun. Chem., 8(1), 281. 10.1038/s42004-025-01667-z
- Neha, Tiwari, V., Mondal, S., Kumari, N., & Karmakar, T. (2022). Collective Variables for Crystallization Simulations- from Early Developments to Recent Advances. ACS Omega, 8(1), 127–146. 10.1021/acsomega.2c06310
- Garcı́a, A. E. (1992). Large-Amplitude Nonlinear Motions in Proteins. Phys. Rev. Lett., 68(17), 2696. 10.1103/physrevlett.68.2696
- Amadei, A., Linssen, A. B., & Berendsen, H. J. (1993). Essential Dynamics of Proteins. Proteins Struct Func Bioinf, 17(4), 412–425. 10.1002/prot.340170408
- Hegger, R., Altis, A., Nguyen, P. H., & Stock, G. (2007). How Complex Is the Dynamics of Peptide Folding? Phys. Rev. Lett., 98(2), 028102. 10.1103/physrevlett.98.028102
- Wetzel, S. J. (2017). Unsupervised Learning of Phase Transitions: From Principal Component Analysis to Variational Autoencoders. Phys. Rev. E, 96(2), 022140. 10.1103/physreve.96.022140
- Ferguson, A. L., Panagiotopoulos, A. Z., Debenedetti, P. G., & Kevrekidis, I. G. (2011). Integrating Diffusion Maps with Umbrella Sampling: Application to Alanine Dipeptide. J. Chem. Phys., 134(13). 10.1063/1.3574394
- Preto, J., & Clementi, C. (2014). Fast Recovery of Free Energy Landscapes via Diffusion-Map-Directed Molecular Dynamics. Phys. Chem. Chem. Phys., 16(36), 19181–19191. 10.1039/c3cp54520b
- Ceriotti, M., Tribello, G. A., & Parrinello, M. (2011). Simplifying the Representation of Complex Free-Energy Landscapes Using Sketch-Map. Proc. Nat. Acad. Sci., 108(32), 13023–13028. 10.1073/pnas.1108486108
- Tribello, G. A., Ceriotti, M., & Parrinello, M. (2012). Using Sketch-Map Coordinates to Analyze and Bias Molecular Dynamics Simulations. Proc. Nat. Acad. Sci., 109(14), 5196–5201. 10.1073/pnas.1201152109
- Chen, W., & Ferguson, A. L. (2018). Molecular Enhanced Sampling with Autoencoders: On-the-fly Collective Variable Discovery and Accelerated Free Energy Landscape Exploration. J. Comput. Chem., 39(25), 2079–2102. 10.1002/jcc.25520